Downloadable R Script · 2,190+ lines · Fully commented
Complete 16S
R Code for
Downstream Analysis
A complete, annotated R script covering the full 16S downstream analysis pipeline — from QIIME2 or CSV to 33+ figures, statistical tables, and a paste-ready Methods section.
Get instant access — $79.00
See what's inside ↓
One-time purchase · RStudio ready
$79.00
13analysis sections
33+figures produced
5CSV exports
3vars to configure
You get the .r source file
QIIME2 .qza or plain CSV
phyloseq · vegan · ggplot2
Fully commented

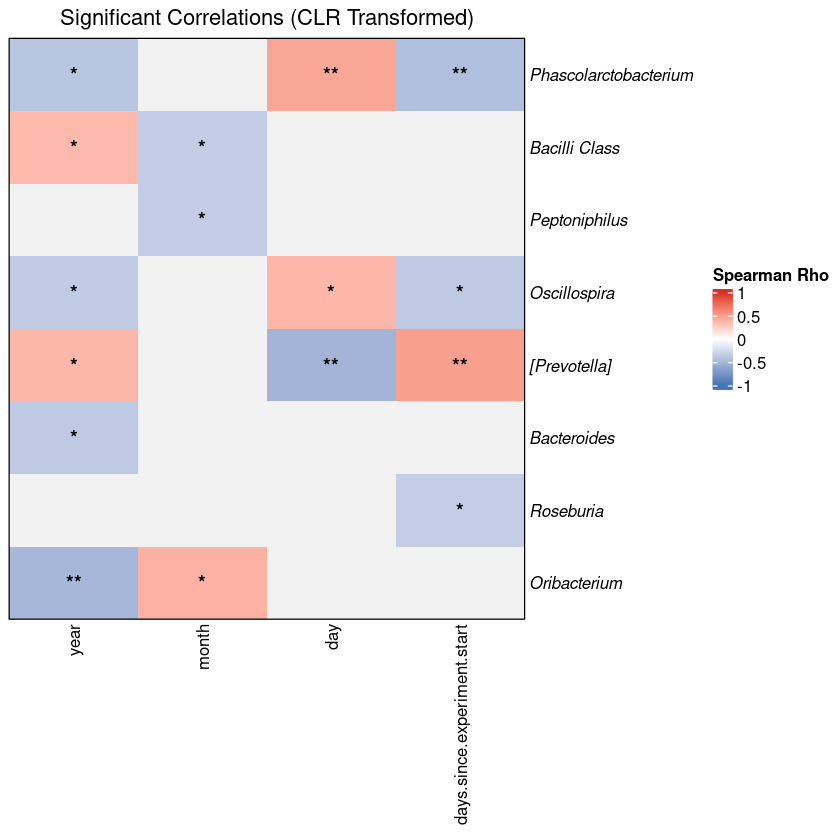
live demoThe script running in RStudio — figures appear section by section
# Set these 3 variables, then run sections 0 → 13 MY_WORKING_DIR <- "path/to/your/project" MY_SAMPLE_ID <- "sample_name" MY_GROUP_VAR <- "Treatment" # QIIME2 .qza — or plain CSV